Toolkit#
Preparing for analysis#
Input file types#
modality XPLR offers a variety of analysis workflows, that leverage common input files and parameters.
The main input files are described in this section, and include:
Zarr store files,
.zarrzSample sheet,
.csvor.tsvRegion files in BED3+ format,
.bedor.tsv
Input files from external sources may be required, such as annotation references (.gtf or .gff).
Descriptions of these file types and formats are also provided below.
Zarr store#
The Zarr store is a compressed file format (.zarrz) used to store large datasets in a
distributed manner. It is designed for efficient storage and retrieval
of large arrays, making it suitable for high-throughput sequencing data
such as methylation data. The Zarr format allows for chunked storage,
enabling efficient access to subsets of data without loading the entire
dataset into memory. This is particularly useful for large datasets
that may not fit into memory all at once.
The Zarr store is a directory structure that contains metadata and data files. The metadata files describe the structure and properties of the dataset, while the data files contain the actual data. The Zarr store can be accessed using various programming languages and tools, including Python, R, and command-line interfaces.
In the context of modality XPLR, the Zarr store contains methylation count data for one of the cytosine contexts: CpG, CHG, or CHH. By default, the upstream duet pipeline generates CpG context data, but it can be configured to produce CHG or CHH context data as well. Each Zarr store contains data for only one context type at a time. For each site in the selected context, the Zarr store records the number of modified cytosines (5-methylcytosine and 5-hydroxymethylcytosine for duet evoC, modified cytosines for duet +modC) and unmodified cytosines, as identified in base-resolution sequencing data, along with total coverage and related counts (see table below for a full list).
For biomodal workflows, the Zarr store containing methylation data is produced by the upstream duet software.
You will need the path to each Zarr store for your dataset to run the modality XPLR software.
For simplified management, Zarr stores can be combined. See the section for Handling Zarr stores and the modality zarr-utils join operation for more information.
A list of count data variables contained in the Zarr store is provided below:
Data variable |
Description |
|---|---|
|
Unmodified C’s. |
|
5-Methylcytosine (5mC) present in 6-base Zarr |
|
5-Hydroxymethylcytosine (5hmC) present in 6-base Zarr. |
|
Combined 5mC and 5hmC signal (modC). |
|
Total number of modified and unmodified C’s
( |
|
Sum of |
|
SNPs in CpGs, i.e. where the CpG base is not a |
Sample sheet#
The sample sheet is a .csv or .tsv file that contains metadata about the samples
in the Zarr store. The sample sheet can be created using any text editor. It must be saved as a .csv or .tsv file with the header row included.
The sample sheet must include a column header, sample_ids, listing one or more sample IDs that match the sample IDs in the Zarr
store. The sample sheet can also include additional columns with
metadata, such as grouping information (e.g., sample type, condition), or covariates (e.g., age, smoker status, batch information).
The analysis operations can handle both continuous and categorical metadata.
Formatting the sample sheet
modality XPLR expects UTF-8 encoding for sample sheet files but is fully compatible with ASCII characters. Do not use non-ASCII (Unicode) characters.
Avoid using special characters, spaces, or commas in the sample sheet. We recommend using underscores _ instead of spaces in the sample IDs and column headers.
If you edit the sample sheet in a text editor, ensure it is saved as plain .csv or .tsv and that no extra spaces or formatting are introduced.
Column Headers
Column headers must not contain spaces or commas.
Use only ASCII letters, numbers, and underscores
_in column headers.Each column header must be unique.
The first column must be named
sample_id(case-sensitive) and contain unique sample identifiers.Additional columns can be used for grouping e.g.,
disease_stageor covariates e.g.,age,smoker_status.
Sample IDs
Sample IDs must not contain spaces or commas.
Use only ASCII letters, numbers, and underscores
_in sample IDs.Each sample ID must be unique within the sample sheet.
Sample IDs must exactly match those used in the Zarr store(s).
Important
Sample sheet metadata can be used for grouping samples in the analysis. Ensure that one of your columns can be used to group samples
to use the grouping functions --aggregate-by-group for Biological QC, DMR calling and Feature Extraction.
The sample sheet should be formatted as follows:
e.g., Sample IDs to be grouped by disease_stage, with the covariate smoker_status included in the analysis.
sample_id |
disease_stage |
smoker_status |
|---|---|---|
sample1 |
control |
no |
sample2 |
I |
yes |
sample3 |
I |
no |
sample4 |
IV |
yes |
sample5 |
IV |
no |
Define a single region#
Several of the tools in the modality XPLR software allow you to
specify a single region of interest for analysis. This can be done by
specifying the region in the command line using the format
chr or chr:<start>-<end>. The start and end positions are optional, and
indices are zero-based and half-open.
For example, to specify the region on chromosome 1 from position 1000 to 2000, use the
following format: chr1:1000-2000. This will restrict the
analysis to a specific region of the genome, which can be useful for
focusing on regions of interest or reducing the size of the
dataset during analysis. The region can be specified in the command line
using the --region option. For example:
modality dmr call \
--zarr-path path/to/data.zarrz \
--region chr1:1000-2000 \
--output-dir path/to/output
Note
If using --region in combination with --window-size, the specified region will be divided into
windows of the specified size. Similarly, you can use a bedfile format to specify individual window sizes over a larger
region of interest. See the Region files section below.
Region files#
The user-supplied region file is a BED3+ format (.bed or .tsv) file that contains genomic regions of interest
for analysis. It should include columns for the chromosome, start
position, and end position of each region. The region file is optional
but can be used to restrict the analysis to specific regions of the
genome. The region file should be formatted as follows:
e.g., Region file with regions of interest.
chromosome |
start |
end |
|---|---|---|
chr1 |
1000 |
2000 |
chr1 |
3000 |
4000 |
chr2 |
5000 |
1000 |
chr2 |
7000 |
3000 |
Annotations are optional but can be included in the analysis to provide additional context and information about the genomic regions of interest. Annotations can include information such as gene names, gene symbols, gene biotypes, and other relevant features. The annotations must be included in the region file and should be formatted as follows:
e.g., Region file with annotations.
chromosome |
start |
end |
annotation |
name |
|---|---|---|---|---|
chr4 |
25250935 |
25251935 |
Promoter |
KRAS |
chr5 |
112706517 |
112707517 |
Promoter |
APC |
chr17 |
7687537 |
7688537 |
Promoter |
TP53 |
Note
A region file can contain multiple different annotation classes, such as promoters, enhancers, or other genomic features.
Preparing annotation files#
There are two types of annotation files that are used in the modality XPLR software:
Annotated region files: Used to define genomic regions for analysis. E.g. as input to the
modality dmr callandmodality gettools.Genome annotation GTF/GFF3 files: Used to annotate new region definitions with
modality prepare-regions(GFF3) or to place analysis outputs into a broader biological context with themodality tracks(GFF/GTF) tool.
You can download the human hg38 GENCODE v44 annotation files for the human (hg38) genome from biomodal, or prepare your own annotation files from public databases as described below.
To access biomodal demo data resouces, you need to install gcloud CLI and authenticate. See the download instructions here then use the following command:
gcloud storage cp gs://biomodal-data/annotation/gencode.v44.basic.annotation.gff3.gz .
gcloud storage cp gs://biomodal-data/annotation/gencode.v44.annotation.gtf.gz .
Downloading directly from GENCODE#
Annotation files can be downloaded directly from the GENCODE website for human or mouse genomes. GENCODE offers several annotation contents and region sets. The table below summarises the options most relevant to modality XPLR:
Content |
Region |
Description |
|---|---|---|
Comprehensive gene annotation |
PRI |
Full gene definitions including all known transcripts, promoters, and exon structures. Includes protein-coding genes, lncRNAs, and other commonly used gene types. Recommended for most users. |
Basic gene annotation |
PRI |
One representative model per gene with minimal feature overlap. Fewer alternative transcripts; faster for gene-level aggregation, but less biologically detailed than Comprehensive. |
Note
Region sets: CHR includes reference chromosomes only; PRI (Primary assembly) covers chromosomes and scaffolds; ALL additionally includes assembly patches and alternate loci (haplotypes). PRI is sufficient for most analyses.
What to download
If you are unsure which file to use, download the Comprehensive gene annotation – PRI (either .gff3.gz or .gtf.gz).
This provides full and up-to-date gene definitions and works cleanly with standard GRCh38 (human) or GRCm38/GRCm39 (mouse) genomes.
Use the Basic gene annotation – PRI if you prefer simpler gene-level summaries with one representative model per gene and less overlap between features.
Alternatively, annotation files can be downloaded from e.g., the UCSC genome browser.
The UCSC has a powerful Table Browser tool, that can be used to download annotation files for different
reference genomes, either as complete files or as subsets of the genome. The output file type can be specified (.gtf or .bed) according to the utility you require.
The v44 human annotation above, was generated using the following steps:
Go to the UCSC Table Browser.
Select the following options:
clade: Mammal
genome: Human
assembly: hg38
group: Genes and Gene Predictions
track: Choose GENCODE v44
table: e.g., refGene, knownGene, ensGene, ncbiRefSeqGene
region: genome
output format: e.g. GTF or BED
output filename: e.g., gencode.v44.annotation.gtf or gencode.v44.basic.annotation.gff3
Click on the “get output” button to download the annotation data.
You are also able to perform additional customisation of the output e.g., of the track header or the rows in your BED output or select using the default settings.
When you are ready, click on the “get output” button again and save the file on your local machine.
Note the file path for use in
modalitycommands.
CpG island annotations can be downloaded from the UCSC Table Browser.
Go to the UCSC Table Browser and select the following options:
Option
Human (hg38)
Mouse (mm10 / mm39)
genome
Human
Mouse
assembly
hg38
mm10 (or mm39)
group
Regulation
Expression and Regulation
track
CpG Islands
CpG Islands
table
cpgIslandExt
cpgIslandExt
region
genome
genome
format
BED
BED
Click “get output”, customise the file header if needed (this can be left unticked), then save the file.
Note the file path for use in
modalitycommands.
Pre-prepared BED files#
The modality XPLR software includes a set of pre-prepared BED files for common genomic regions of interest. These .bed.gz files were prepared using:
GENCODE v44 annotation for the human genome (hg38)
GENCODE vM25 annotation for the mouse genome (mm10) - without “chr” prefix
GENCODE vM25 annotation for the mouse genome (mm10p6)
GENCODE M33 annotation for the mouse genome (mm39) - without “chr” prefix
The available region files are:
Genes
Promoters
TSS
CpG Islands
CpG Shores
CpG Shelves
These files can be used as input to the modality dmr call and modality get tools to restrict the analysis to specific regions of the genome.
Contact support@biomodal.com or use the links below to obtain these files.
Download human hg38 Annotated BED Files.zip
Download mouse mm10 Annotated BED Files.zip
Download mouse mm10p6 Annotated BED Files.zip
Download mouse mm39 Annotated BED Files.zip
How were these files created?
Genes: Read-through transcripts were removed from the GENCODE annotation file (i.e., transcripts with exons spanning >1 gene). This is because:
The genes involved are found on the same chromosome region on the same strand, typically adjacent to one another.
Their role is unclear
It’s common to find them spanning up to 2 genes, but can span >2
They are ubiquitous
Promoters: Promoters were defined as the 1kb region upstream of the gene
startposition and are 1000bp in length.This is considered a good estimate and not a golden rule for all genes, as promoter distances from the 5’UTR will differ.
Because the gene-level start coordinate is used, this approach captures only one promoter per gene. Genes with well-characterised alternative promoters driving different isoforms (e.g., many genes in cancer-relevant pathways) will have those isoform-specific regulatory regions excluded. If your analysis is specifically focused on isoform-level regulation, consider supplementing with a custom BED file that defines the individual transcript start sites of interest.
TSS: Transcription start sites (TSS) were defined as the 200bp region upstream of the gene
startposition and are 400bp in length.
Note
Whilst genes may have multiple transcripts, we have used the GENCODE main gene definition to define a single TSS and promoter per gene for these files.
CpG shores and shelves files were created from the list of CpG islands in the GENCODE annotation file, using the following definitions:
CpG Shores: CpG shores were defined as the 2kb region upstream and downstream of the CpG island.
CpG Shelves: CpG shelves were defined as the 2kb region upstream and downstream of the CpG shores.
Note
When two CpG islands are closer than the extension distance for shores and shelves (less than 4 kb for shores or less than 8 kb for shelves), the extended regions will overlap. In such cases, the overlapping segments are merged into a single continuous region.

Figure 3 Definitions of CpG Islands, Shores and Shelves in pre-prepared BED files.#
Top up sequencing & aggregating samples#
When samples are sequenced across multiple sequencing runs (top-up sequencing), you can aggregate the data from these runs during analysis. modality XPLR handles multi-run data through analysis-time aggregation, where samples are combined at the point of running analysis.
Analysis-time aggregation workflow
Sample aggregation is performed during analysis execution and requires:
Pre-joining Zarr stores: For analyses with top-up sequenced samples in separate runs, we reccommend to join the Zarr stores before analysis. Use the
modality zarr-utils joincommand to merge multiple Zarr stores. The sample IDs must be unique, with technical replicates differentiated with e.g._rep1,_rep2. See Handling Zarr stores for detailed instructions. Once joined, samples are handled during analysis using the methods described below.
Note
If samples need to be renamed prior to joining, see Renaming samples for detailed instructions.
Sample sheet preparation: Create a sample sheet with a column that groups matched samples from different runs using a common identifier (e.g., the sample ID without the
_rep*suffix for technical replicates).Analysis-specific handling: Use the appropriate parameter for your chosen analysis tool:
Operation |
Analysis tool. |
Function |
|---|---|---|
–aggregate-by-group <label> |
|
Performs aggregation by summing count values in the Zarr store by a common metadata <label>, before analysis. Result files (BED3+) are reduced and plots show aggregated results. |
–condition-array-name <label> |
|
Does not perform aggregation. Defines DMR groups by metadata <label> prior to DMR calling.
Technical replicates are considered as separate samples during DMR calling, even if they are assigned the same
group label ( |
–order-by-group <label> |
|
Does not perform aggregation. Use this feature to order samples by metadata <label>, to enhance visual order in plots. Result (BED3+) files are not ordered or reduced by metadata <label>. |
Example workflow for top-up sequencing:
If you have sample “Patient_001” sequenced in two different runs (Run_A and Run_B), your sample sheet might look like:
sample_id |
patient_id |
condition |
|---|---|---|
Patient_001_Run_A |
Patient_001 |
control |
Patient_001_Run_B |
Patient_001 |
control |
Patient_002_Run_A |
Patient_002 |
treatment |
Patient_002_Run_B |
Patient_002 |
treatment |
For modality biological-qc or modality get, use --aggregate-by-group patient_id to combine data from both runs for each patient. For modality dmr call, use --condition-array-name condition to group samples by treatment condition prior to DMR calling.
Important
When aggregating samples from multiple runs, ensure that:
Sample identifiers in the Zarr stores match those listed in the sample sheet.
The aggregation column in your sample sheet correctly groups samples that should be combined under a common ID.
Run-level metadata is preserved in the sample sheet for quality control purposes.
Note
Aggregation is performed by summing count values for samples within the same group, making it suitable for combining technical replicates or top-up sequencing data.
Merging CpG strands#
By default, modality XPLR merges CpG strands for analysis by summing the counts on the two strands.
This is true for export, biological-qc, dmr call and get. Each entrypoint allows a --disable-strand-merging flag to disable strand merging if desired.
The modality tracks entrypoint is configured by the .ini file that is generated during DMR calling (dmr call), which contains a setting to enable or disable strand merging.
This is because the tracks feature plots a methylation trace directly from the Zarr store data, and separate DMR modification difference and magnitude tracks that are based on DMR results files.
Therefore, the dmr call setting for strand merging is carried over to the .ini so that tracks are drawn on matched data.
Here’s an example of how strand merging works for 5mC (the same logic applies to 5hmC and modC):
If we had a CpG at reference position 10 on the + strand, and the corresponding CpG at position 11 on the - strand,
the total number of C’s may look like:
Position |
Strand |
mC |
Total C |
|---|---|---|---|
10 |
|
8 |
20 |
11 |
|
5 |
15 |
If strands are merged (default), the counts for the two strands are summed.
So the merged CpG will have 8 + 5 = 13 mC, and 20 + 15 = 35 Total C, so its methylation fraction is 13 / 35 = 0.37.
If the --disable-strand-merging flag is used, strand merging is set to False and the counts for the two strands are kept separate.
The CpG at position 10 has a methylation fraction 8 / 20 = 0.4, and the CpG at position 11 has a methylation fraction 5 / 15 = 0.33.
Note
The + strand marks the reference position of the merged CpG, so in this example the merged CpG will be at position 10.
Saving modality XPLR output files#
modality XPLR defaults to saving output files in the current working directory.
To avoid outputs being overwritten, the output files or sub-folders created during analysis are time-stamped.
It is recommended to specify an output directory for each analysis using the command option --output-dir, where applicable.
Handling Zarr stores#
The modality zarr-utils command group provides tools for managing and manipulating Zarr stores used in methylation analysis.
These utilities help you to inspect & rename samples as well as join Zarr stores from different runs for downstream analysis.
Overview#
Zarr stores are the primary data format for modality XPLR workflows. The zarr-utils entrypoint offers subcommands to:
Inspect sample IDs in a Zarr store.
Rename sample IDs (with safety checks).
Generate template mapping files for renaming.
Join Zarr stores from different runs.
These utilities are especially useful when integrating data from multiple sources, standardising sample identifiers, or preparing data for collaborative projects.
Subcommands#
The subcommands are:
rename-sampleswhich allows you to view and modify sample IDs in a Zarr store.joinwhich allows you to combine multiple Zarr stores into a single store.
Renaming samples#
This command lets you list and rename sample IDs in your Zarr store. The sample renaming can either be done directly by passing a command to define current and new values,
or by passing a .csv or .json file for batch processing. You can generate a template .csv or .json file with your samples pre-populated for easier renaming of multiple samples at the same time.
Important
After renaming samples in a Zarr store, you must manually update the metadata (sample sheet) file to reflect the new sample IDs.
Inputs#
modality zarr-utils rename-samples requires the following inputs:
Zarr store: Path to a Zarr store, with samples to be renamed.
Mapping: Either a
.csvor.jsonfile containing the mapping of current sample IDs to new sample IDs, or an inline mapping string (e.g.,old1:new1,old2:new2).
Command and outputs#
The rename-samples tool is invoked by typing modality zarr-utils rename-samples into the CLI terminal. To display a list of permitted arguments in the terminal, enter:
modality zarr-utils rename-samples --help
Typical usage examples:
modality zarr-utils rename-samples \
--zarr-path <path/to/data.zarrz> \
--mapping <path/to/sample_mapping.csv> \
--output-path <path/to/renamed_data.zarrz>
# To overwrite the original store (use with caution)
modality zarr-utils rename-samples \
--zarr-path <path/to/data.zarrz> \
--mapping <path/to/sample_mapping.csv> \
--overwrite
The following table describes the tool options:
OPTION |
Required |
Description |
|---|---|---|
|
Yes |
Path to the Zarr store to inspect or modify. |
|
No |
Mapping of current to new sample IDs. Accepts a |
|
No |
Path to save the modified Zarr store. If not provided, the original store will be overwritten (requires –overwrite). |
|
No |
Allow overwriting the original Zarr store. Default: FALSE. |
|
No |
Display current sample IDs and exit without making changes. Default: FALSE. |
|
No |
Generate a template mapping file ( The headers are set as |
Analysis examples#
Example 1: List sample IDs in a Zarr store#
modality zarr-utils rename-samples \
--zarr-path <path/to/data.zarrz> \
--list-samples
Example 2: Generate a template mapping file for renaming#
modality zarr-utils rename-samples \
--zarr-path <path/to/data.zarrz> \
--generate-template <path/to/sample_mapping.csv>
Edit the generated template file to specify new sample IDs, then use it with --mapping.
Example 3: Rename sample IDs using a mapping file#
modality zarr-utils rename-samples \
--zarr-path <path/to/data.zarrz> \
--mapping <path/to/sample_mapping.csv> \
--output-path <path/to/renamed_data.zarrz>
Example 4: Rename and overwrite the original store (use with caution)#
modality zarr-utils rename-samples \
--zarr-path <path/to/data.zarrz> \
--mapping <path/to/sample_mapping.csv> \
--overwrite
Important
Safety and Best Practices:
By default, the tool will not overwrite the original Zarr store unless
--overwriteis explicitly set.If you do not specify
--output-path, you must use--overwriteto confirm you want to modify the original file.The tool checks for conflicts (e.g., new sample IDs that already exist) and will raise an error if detected.
Always review the list of sample IDs before and after renaming to ensure correctness.
Note
For more details on Zarr store structure and sample sheet requirements, see the Zarr store section above.
Combining Zarr store files#
The modality XPLR software allows you to combine multiple Zarr
stores into a single Zarr store for analysis, using the modality zarr-utils join command.
Important
Joining 5-base (duet +modC) and 6-base (duet evoC) Zarr stores is not supported.
Caution should be taken when joining Zarr stores between duet pipeline versions where there could be significant changes impacting methylation quantification.
Joining Zarr stores using modality zarr-utils join is ideal for:
Organising data from multiple sequencing runs.
Avoiding entering multiple Zarr paths in the command line.
Where applicable, run-level information for each sample should be recorded in the metadata --sample-sheet for downstream analyses, such as the modality dmr call workflow.
Inputs#
modality zarr-utils join requires the following inputs:
Zarr store(s): Paths to each Zarr store to be combined.
Output directory and filename: Full path to the new compressed output file
.zarrzcontaining the combined data.
Tip
To specify all Zarr stores in a single directory, you can use a wildcard pattern in a single path argument. E.g. path/to/directory/*.zarrz.
Command and outputs#
The combine tool is invoked by typing modality zarr-utils join into the CLI terminal. To display a list of permitted arguments in the terminal, enter:
modality zarr-utils join --help
Typical usage example:
modality zarr-utils join \
--zarr-path <path/to/data1.zarrz> <path/to/data2.zarrz> \
--output-path <path/to/new/combined.zarrz> \
The following table describes the tool options:
OPTION |
Required |
Description |
|---|---|---|
|
Yes |
Two or more Zarr files to be combined. e.g., |
|
Yes |
Full path to an output combined zipped
e.g., |
Exporting Zarr stores to community-supported file types#
The modality export tool allows you to export methylation data stored in Zarr format to widely used file formats.
This functionality is essential for integrating data with other bioinformatics tools, sharing data with collaborators, or preparing data for publication.
The supported export formats are:
File type |
CLI option |
Description |
IGV compatible |
|---|---|---|---|
BedGraph |
|
Export data to BedGraph format. |
Yes |
BedMethyl |
|
Export data to BedMethyl format. |
Yes |
Bismark |
|
Export data to Bismark format. |
Yes |
CX-report |
|
Export data to CX-report format. |
No |
Quant |
|
Export data to quant file containing methylation counts on a per CpG basis. |
No |
The tool provides flexibility in selecting specific samples, regions, and data fields for export, ensuring compatibility with downstream workflows.
Inputs#
modality export requires you to provide an export/file type e.g., bismark or bedgraph and has 1 mandatory input.
Zarr store: Path to a Zarr store to be exported
This will however export all of your samples to the selected file type, if you want to export only a subset of your samples, you can use the --sample-ids option to specify a list of sample IDs to export. You can list the sample IDs in your Zarr store using the zarr-utils command, for more details, see Example 1: List sample IDs in a Zarr store in Renaming samples.
Command#
The export tool is invoked by typing modality export into the CLI terminal. To display a list of permitted options and export formats, enter:
modality export --help
To display a list of permitted arguments for a specific export format, enter:
modality export <export-format> --help
# e.g., for Bismark
modality export bismark --help
# e.g., for BedGraph
modality export bedgraph --help
# e.g., for BedMethyl
modality export bedmethyl --help
# e.g., for CX-report
modality export cxreport --help
# e.g., for Quant
modality export quant --help
Typical usage example:
modality export bismark \
--zarr-path <path/to/data.zarrz> \
--tag genome \
--modification-count-column modc \
--denominator-column total_c \
--skip-rows-zero-counts \
--output-dir <path/to/output>
The following table describes the tool arguments for the export options:
OPTION |
Required |
Description |
|---|---|---|
|
Yes |
Path to the Zarr file to be exported. e.g., |
|
No |
List of sample ID strings to include in the export. If not specified, all samples are included. One export file is produced per sample. e.g., |
|
No |
Tag string to append to the output file name.
The output file name will use the
format:
e.g., |
|
No |
Specify the column used for methylation counts.
See Zarr store for a list of available data columns.
Default: e.g., |
|
No |
Specify the column used to calculate
the methylation fraction and coverage.
See Zarr store for a list of available data columns.
Default: e.g., |
|
No |
Specify additional columns to include in the export. See Zarr store for a list of available data columns. e.g., |
|
No |
Integer to specify the minimum context sequencing depth for filtering. Filters the positions if the mean coverage across all samples in each position is lower than the desired filter value. e.g., |
|
No |
Flag to disable strand merging to retain separate CpG sites on opposite strands (default: False, strands are merged). e.g., |
|
No |
Flag to turn ON, skip rows with zero counts. e.g., |
|
No |
Compress output files using bgzip and create Tabix index. This option is only available for BedGraph and BedMethyl formats. Requires installation of non-python dependencies (see installation section). e.g., |
|
No |
Directory to save the exported files. Default: current working directory. e.g., |
|
No |
Restrict export to a specific genome
region. Use the format e.g., |
For more information on strand merging, see the Merging CpG strands section.
Outputs#
The modality export tool generates files in the specified format, saved in the output directory. The file names include the sample ID, tag, and data type. For example:
Bismark:
sample1.genome.modc_bismark.bed.gzCX-report:
sample1.genome.modc_cxreport.txt.gzBedGraph:
sample1.genome.modc_bedgraph.gzBedMethyl:
sample1.genome.modc_bedmethyl.gzQuant:
sample1.genome.modc_quant.tsv.gz
If --tabix is specified, an additional .gz.tbi output will be created per sample.
An additional provenance file is created in the output directory, named modality_metadata_<YYYYMMDD>_<HHMMSS>.json, which contains metadata about the export process, including the command used, input files, and parameters.
Example 1: Export bismark coverage report#
modality export bismark \
--zarr-path <path/to/data.zarrz> \
--tag genome \
--modification-count-column modc \
--denominator-column total_c \
--skip-rows-zero-counts \
--tabix \
--output-dir <path/to/output>
Example 2: Export bedgraph for specific region#
modality export bedgraph \
--zarr-path <path/to/data.zarrz> \
--region chr1:1000-2000 \
--output-dir <path/to/output>
Example 3: Export quant format for selected samples#
modality export quant \
--zarr-path <path/to/data.zarrz> \
--sample-ids sample1 sample2 \
--tag genome \
--output-dir <path/to/output>
Prepare annotated regions#
CpG-informed segmentation for region generation#
The modality prepare-regions command generates and optionally annotates genomic regions from a Zarr dataset, using a CpG-informed segmentation
approach. The bedfile output can then be used in downstream workflows such as modality get and modality dmr call, as with using Pre-prepared BED files.
Instead of using fixed window sizes, CpG-informed segmentation defines regions based on the distribution of CpG sites in the genome.
CpG-informed segmentation works by identifying clusters of CpG sites and segmenting the genome between them. New segment definitions are
based on user-controlled settings e.g., maximum region size, minimum number of CpG contexts per region, and a threshold for
splitting continuous CpG positions into separate segments. A new segment starts when the gap between consecutive positions exceeds the provided value. Finally, each
of these segments/regions can be annotated with features from a GFF3 file and/or CpG island annotation file. For transcript-level features (exons, introns, 5’ UTRs and 3’ UTRs), canonical transcripts are used by default;
users can toggle this off and use all transcript isoforms by setting the flag --include-all-transcripts. Promoter and gene body annotations are
always derived from the GENCODE gene-level record, which defines a single coordinate span per gene regardless of isoforms, and are therefore
unaffected by --include-all-transcripts.
Inputs#
modality prepare-regions requires the following input:
Zarr store(s): One or more paths to Zarr files containing methylation data.
The following inputs are optional inputs used for region generation and annotation. In the case of annotation files, one or multiple types may be provided.
- Region generation
CpG segmented region max size: Maximum size (in base pairs) for CpG-segmented regions
CpG segmented split segment threshold: Threshold for splitting contiguous CpG positions
CpG min contexts: Minimum number of CpG contexts required per region.
- Region Annotation
GFF3 annotation file: Path to a GFF3 annotation file used to annotate generated regions.
CpG island annotation file: Path to a CpG island annotation file used for region annotation, e.g. downloaded from UCSC.
Minimum overlap: Minimum number of bases or fraction of bases required to overlap for successful annotation.
Use canonical transcripts for annotation (default): For transcript-level features (exons, introns, 5’ UTRs and 3’ UTRs), only the canonical transcript isoform is used. Users can toggle this mode off and include all transcript isoforms by setting the flag
--include-all-transcripts. This flag has no effect on promoter or gene body annotations, which are always derived from the single gene-level GENCODE record.The canonical transcript is selected per gene using the following priority order:
MANE Select — the clinically relevant transcript jointly defined by NCBI and ENSEMBL (highest priority).
APPRIS Principal — APPRIS principal isoform level (
appris_principal_1>appris_principal_2> …).Ensembl canonical — the transcript flagged as canonical by Ensembl.
Longest transcript — used as a fallback if none of the above tags are present.
These tags are read directly from the
tagattribute of the GFF3 file. If none of MANE, APRIS principal or Ensembl canonical annotations are present, the fallback will be the longest transcript. Note that, if you use a custom GFF3 that does not contain any of these tags, the fallback will also be the longest transcript.
Note
Promoter annotations always produce one region per gene, regardless of the number of transcript isoforms. This is because they are derived from the GFF3 gene feature, which defines a single coordinate span for the whole gene. The promoter is defined as the 1 kb region upstream of the gene-level start position (strand-aware). Isoform-specific alternative promoters are not represented and --include-all-transcripts does not affect this behaviour. See the discussion of this limitation in Pre-prepared BED files.
Downloading annotation files#
It is reccommended the GFF3 and CpG Island annotation files are downloaded from UCSC, according to your required parameters. See Preparing annotation files for more details.
Command#
OPTION |
Required |
Description |
|---|---|---|
|
No |
A Zarr file containing methylation data. e.g., |
|
No |
Path to a GFF3 annotation file used to annotate generated regions. |
|
No |
Path to a CpG island annotation file (BED) used for region annotation. |
|
No |
Maximum region size (in base pairs) for CpG-segmented regions. |
|
No |
Threshold for splitting contiguous CpG positions into separate segments. A new segment starts when the gap between consecutive positions exceeds this value. |
|
No |
Minimum number of CpG contexts required per region. |
|
No |
Minimum number of bases or fraction of bases required to overlap for successful annotation. |
|
No |
Output directory for generated files. Defaults to the current directory. |
|
No |
Select one or more annotation(s) to include in the output. Valid values: |
|
No |
Include all transcript isoforms when annotating exons, introns, 5’ UTRs and 3’ UTRs, instead of only using the canonical transcript. Default: False (canonical transcript only). Note: this flag has no effect on promoter or gene body annotations, which are always derived from the single gene-level GENCODE record. |
|
No |
Show the help message and exit. |
Analysis examples#
Example 1 - Generate regions and annotate with GFF3 using only canonical transcripts#
modality prepare-regions \
--zarr-path <path/to/data.zarrz> \
--gff3-annotation-file <path/to/annotation.gff3> \
--cpg-segmented-region-max-size 5000 \
--cpg-segmented-split-segment-threshold 500 \
--cpg-segmented-min-contexts 5 \
--min-overlap 1 \
--annotate promoter 5utr 3utr exon gene intron \
--output-dir <path/to/output>
Example 2 - Generate regions and annotate all features with GFF3 using all transcripts#
modality prepare-regions \
--zarr-path <path/to/data.zarrz> \
--gff3-annotation-file <path/to/annotation.gff3> \
--cpg-segmented-region-max-size 5000 \
--cpg-segmented-split-segment-threshold 500 \
--cpg-segmented-min-contexts 5 \
--min-overlap 1 \
--annotate all \
--include-all-transcripts \
--output-dir <path/to/output>
Example 3 - Generate regions and annotate canonical features with GFF3 & CpG islands#
modality prepare-regions \
--zarr-path <path/to/data.zarrz> \
--gff3-annotation-file <path/to/annotation.gff3> \
--cpg-island-annotation-file <path/to/cpg_islands.bed> \
--cpg-segmented-region-max-size 5000 \
--cpg-segmented-split-segment-threshold 500 \
--cpg-segmented-min-contexts 5 \
--min-overlap 1 \
--annotate all \
--output-dir <path/to/output>
Example 4 - Generate regions without annotations#
modality prepare-regions \
--zarr-path <path/to/data.zarrz> \
--cpg-segmented-region-max-size 5000 \
--cpg-segmented-split-segment-threshold 500 \
--cpg-segmented-min-contexts 5 \
--min-overlap 1 \
--output-dir <path/to/output>
Outputs#
The modality prepare-regions command produces a BED-formatted file containing the generated genomic regions. These regions are derived using a CpG-based segmentation approach and are annotated using the provided GFF3, CpG island. Each row in the output file represents a single genomic region and includes the following columns:
Chromosome - Chromomsome of the region
Start - Start position of the region
End - End position of the region
Name - Name of the gene (if applicable e.g., not applicable for –annotate cpg_clusters i.e., regions annotated as CpG islands, shelves or shores)
Annotation - Type of feature e.g., promoter, exon, intron etc.
Overlap - Number of bases overlapping with the annotation.
Overlap_fraction - Fraction of the region overlapping with the annotation.
Note
Name, Annotation, Overlap and Overlap_fraction will be delimited by a semicolon ; if multiple region annotations are identified for a single CpG segment. This occurs when a CpG segment overlaps with more than one annotated region, for example:
A CpG segment that falls within both an exon and an intron of different transcripts (when using
--include-all-transcripts).A CpG segment in the shared overlap of two nearby gene promoters. Because promoters are defined as the 1 kb upstream of each gene’s start position, head-to-head gene pairs or closely spaced genes on the same strand can produce overlapping promoter windows. A CpG segment in that shared region will receive both gene names and both promoter annotations, separated by
;.
Biological QC#
Quality control of methylation data#
The biological-qc tool is used to perform biological quality
control (QC) on methylation datasets stored in Zarr format. This command allows
you to ask whether the data makes “biological sense” in the context of a broader experimental design (sample types, replicates, treatment or condition etc.).
Refer to the additional QC metrics generated by the upstream duet software for sequencing-level assessment of individual sample quality.
This command takes input Zarr store(s) and generates an HTML report containing a summary of Zarr and Sample Information, with key visualisations, including Pearson correlation heatmaps and Principal Component Analysis (PCA) scatterplots.
The report provides insights into:
Sample correlations: Identifying relationships between samples to detect outliers or unexpected patterns, or to confirm expected relationships.
Data structure: Using PCA to visualise the variance in the dataset and assess grouping or clustering of samples.
These outputs allow validation of data integrity according to experimental design, prior to downstream analyses such as calling differentially methylated regions (DMRs).
Inputs#
modality biological-qc requires the following inputs:
Zarr store(s): Paths to each Zarr store to include in the analysis.
Sample sheet (recommended): Path to the sample sheet file containing metadata about the samples. Providing this file will allow colour-based grouping controls for the PCA plot on the HTML report.
Output directory (recommended): Path to the output directory where the HTML report will be saved.
Command#
The biological-qc tool is invoked by typing modality biological-qc into the CLI terminal.
To display a list of permitted arguments in the terminal, enter:
modality biological-qc --help
Typical usage examples:
modality biological-qc \
--zarr-path <path/to/data.zarrz> \
--output-dir <path/to/output/directory>
Using shortcuts:
modality biological-qc \
-z <path/to/data.zarrz> \
-otd <path/to/output/directory>
The following table describes the tool options and shortcuts:
OPTION |
Required |
Description |
|---|---|---|
|
Yes |
One or more paths to Zarr files. e.g., |
|
No |
String to restrict analysis to a
specific genome region. Use the
format e.g., |
|
No |
Path to a sample sheet file containing
a column with the header
e.g., |
|
No |
Optional group name for aggregation. Reduces the dataset by summing count values for samples within each group. This affects all downstream analysis (correlation, PCA, etc.). Use this for merging technical replicates or combining samples by condition. Requires |
|
No |
Column name in the sample sheet to use for ordering samples in plots. Samples will be sorted by this group for visual organisation only. Does not perform aggregation - all individual samples remain in the analysis. Requires |
|
No |
Specify which methylation contexts to
analyse. Auto-detects by default:
e.g.:
- |
|
No |
Flag to disable strand merging to retain separate CpG sites on opposite strands (default: False, strands are merged). e.g., |
|
No |
Option to specify a path to an output directory. If not specified, the default output path is to the current working directory. e.g., |
For more information on strand merging, see the Merging CpG strands section.
Outputs#
The modality biological-qc tool generates an HTML report and a provenance metadata file, modality_metadata_<YYYYMMDD>_<HHMMSS>.json, in the output directory.
The HTML report contains the following sections:
Dataset Information
Summary of the dataset, including the reference genome, 5- or 6-base data type (duet +modC or duet evoC), CG/CHG/CHH context, number of samples and number of contexts.
Sample Information
Sample metadata, including Sample IDs and mean CpG coverage that are read from the Zarr store.
If a --sample-sheet metadata file is provided, this table shall also include sample group and covariate information that is read from the sample sheet.
Note
The number of contexts will depend on whether strands are merged (default) or not. Merging strands will show approximately half the number of contexts. The mean CpG coverage also depends on whether strands are merged or not. After merging, the CpG depth will closely match the NGS sequencing depth. If strands are not merged, the average CpG depth will be approximately half the NGS depth, due to a CpG-to-genome-wide coverage ratio of ~0.45-0.50.
Pearson Correlation Heatmap
Visualises the pairwise correlation between samples, helping to identify outliers or unexpected relationships.
Use the --order-by-group option to control the order of samples on the axes based on a specified column in the sample sheet.
Principal Component Analysis (PCA) Plots
Displays the variance in the dataset, showing how samples cluster based on their methylation profiles. Controls to colour the data points by sample group or covariate are provided in the HTML report, allowing you to explore the data in more detail.
Important
Aggregation vs Ordering:
Use
--aggregate-by-groupto reduce samples by summing within groups before all analyses. This creates group-level data (e.g., 3 groups instead of 10 samples) and is useful for technical replicates or combining samples by condition.Use
--order-by-groupto keep all individual samples but organise them by group in visualisations. This does not change the data, only how it’s displayed.
These two options are mutually exclusive - you cannot use both simultaneously. In visualisations, the group order on the axis will follow the list order of the sample sheet.
Note
Methylation Contexts for 6-base Datasets:
By default, 6-base (duet evoC) datasets generate correlation heatmaps and PCA plots for both 5mC and 5hmC separately.
Use --methylation-contexts to customise which contexts to analyse:
--methylation-contexts modcanalyses only the combined modC signal (5mC + 5hmC)--methylation-contexts mcanalyses only 5mC signal--methylation-contexts hmcanalyses only 5hmC signal--methylation-contexts mc hmcanalyses both separately (default behaviour)
For 5-base (duet +modC) datasets, only modC is available and analysed by default.
Analysis examples#
Example 1: Sample-wise QC on a single Zarr dataset#
modality biological-qc \
--zarr-path <path/to/dataset.zarrz> \
--sample-sheet <path/to/metadata.csv> \
--order-by-group <label> \
--output-dir <path/to/output>
Example 2: Ordered sample-wise QC without aggregation#
This example shows how to order samples by a grouping variable without aggregating:
modality biological-qc \
--zarr-path <path/to/dataset1.zarrz> <path/to/dataset2.zarrz> \
--sample-sheet <path/to/metadata.csv> \
--order-by-group <condition> \
--output-dir <path/to/output>
Example 3: Aggregated QC on large datasets from multiple Zarr stores#
This example shows how to aggregate technical replicates:
modality biological-qc \
--zarr-path <path/to/dataset1.zarrz> <path/to/dataset2.zarrz> \
--sample-sheet <path/to/metadata.csv> \
--aggregate-by-group <replicate-group> \
--output-dir <path/to/output>
Calling differentially methylated regions (DMRs)#
Differentially Methylated Regions (DMRs) are genomic regions that exhibit significant differences in DNA methylation levels between at least two different groups.
A region represents the methylation status of summed CpGs within specified genomic coordinates, for each sample.
Identifying DMRs is crucial for understanding the role of DNA methylation in various biological processes, including development, disease, and environmental or treatment responses.
Method used for calling DMRs#
This tool can be used to identify the following types of DMRs, according to the biomodal chemistry used for sample preparation:
modC DMRs: DMRs identified using 5-base (duet +modC)or 6-base (duet evoC) biomodal methylation data, where 5mC and 5hmC signal is combined to a single modC readout.
5mC DMRs: DMRs identified using 6-base biomodal methylation data, where 5mC signal is used to identify DMRs.
5hmC DMRs: DMRs identified using 6-base biomodal methylation data, where 5hmC signal is used to identify DMRs.
Important
The modality dmr call tool is compatible with Zarr stores that
contain either 5-base or 6-base methylation data. See Zarr store for more details on the available fields in the Zarr store.
There are 6 steps in the method for calling DMRs:
Data preparation
Input data consists of methylation counts (e.g., 5mC, 5hmC, modC) stored in a Zarr store, with sample metadata provided in a
--sample-sheet.Optionally, regions of interest can be defined using a BED file or by specifying a
--window-sizefor genome-wide tiling.
Summing counts
For each region (window or BED-defined), modality XPLR sums the counts of methylated and unmethylated cytosines within each region or window.
This produces, for each region and sample, the number of modified (e.g.,
modc) and total cytosines (total_c).
Grouping samples
Samples are grouped according to the experimental design (e.g., disease vs. control), as specified by
--condition-array-name, which matches a column in the sample sheet.
Statistical modeling
For each region, modality XPLR fits a logistic regression model to test for differential methylation between groups.
The model compares the proportion of methylated cytosines between groups, adjusting for
--covariatesif specified and present in the provided sample sheet.The null hypothesis is that there is no difference in methylation between groups.
Wald test
A Wald test is performed on the group coefficient from the logistic regression.
This yields a test statistic and a p-value for each region, indicating the likelihood that observed differences are due to chance.
Multiple testing correction
p-values indicate the probability of observing the data (or something more extreme) under the null hypothesis to test the likelihood that what you are observing is not occurring by random chance.
p-values are adjusted for multiple testing using the Benjamini-Hochberg False Discovery Rate (FDR) method, resulting in a q-value (
dmr_qvalue) for each region.q-values are adjusted p-values that represent the minimum FDR at which a test may be called significant.
Multiple testing correction is necessary when many statistical tests are performed simultaneously, to control for false positives.
See Benjamini-Hochberg correction for more details on the Benjamini-Hochberg correction.
The output of this analysis is a tab-delimited DMR result file adhering to the BED3+ format, for each group pair, and each methylation context that is specified in the command.
In addition, the tool generates an additional .ini file per DMR result file, containing the parameters used for the analysis. The .ini file
records the essential information required to view your DMRs visually in genomic context, including with genetic annotations using the plotting functionality modality tracks, see Generate genomic track plots for more details.
Typical DMR workflow#
The Differentially Methylated Region (DMR) workflow analyses the methylation data to identify regions of the genome that are differentially methylated between experimental groups. The workflow is designed to be flexible and can be applied to a variety of experimental designs, including time-course experiments, treatment vs. control comparisons, and disease vs. normal comparisons.
DMRs can be used to identify hyper-methylated (increase in methylation compared to control) and hypo-methylated (loss of methylation compared to control) regions of the genome,
which can provide insights into the underlying biological processes and mechanisms involved in the experimental conditions being studied.
The direction of methylation change (hyper- or hypo-methylation) is described as the mod-difference.
The DMR workflow includes tools for visualising the data with key statistical metrics for hypotheses to be tested or patterns of methylation to be revealed between different experimental groups. The result data can then be combined with genome annotation tracks to provide biological context to the results.
A typical DMR workflow uses three modality tools:
modality dmr callidentifies the statistically significant DMR(s)modality dmr plotsupports result filtering, QC, and visualisation of hypo- and hyper-methylated regions.modality tracksfor region-specific visualisation of DMRs, with annotated genome context.
This example demonstrates a complete workflow for identifying and visualising DMRs.
Call DMRs:
modality dmr call \
--sample-sheet <path/to/metadata.csv> \
--zarr-path <path/to/data.zarrz> \
--condition-array-name disease_stage \
--methylation-contexts mc hmc \
--window-size 1000 \
--output-dir <path/to/output>
Plot DMR results:
modality dmr plot \
--dmr-results <path/to/output/dmr_results.bed> \
--filter-qvalue 0.05 \
--filter-mod-difference 0.1 \
--output-dir <path/to/output>
Generate genomic track plots:
modality tracks \
--tracks-file <path/to/output/dmr_results.ini> \
--region chr1:100000-200000 \
--output-dir <path/to/output>
The above three sections describe how to call DMRs, how to plot them and how to interpret them for insight into patterns and trends in DNA methylation.
Benjamini-Hochberg correction#
The Benjamini-Hochberg (BH) procedure is a statistical method used to control the false discovery rate (FDR) when performing multiple hypothesis tests. One effect of BH correction, is that multiple DMRs may share the same q-value, particularly when the number of tests is large and the p-values are not uniformly distributed. This is because the BH procedure adjusts p-values based on their rank among all tests, and when many tests yield similar p-values, they may be assigned the same adjusted q-value.
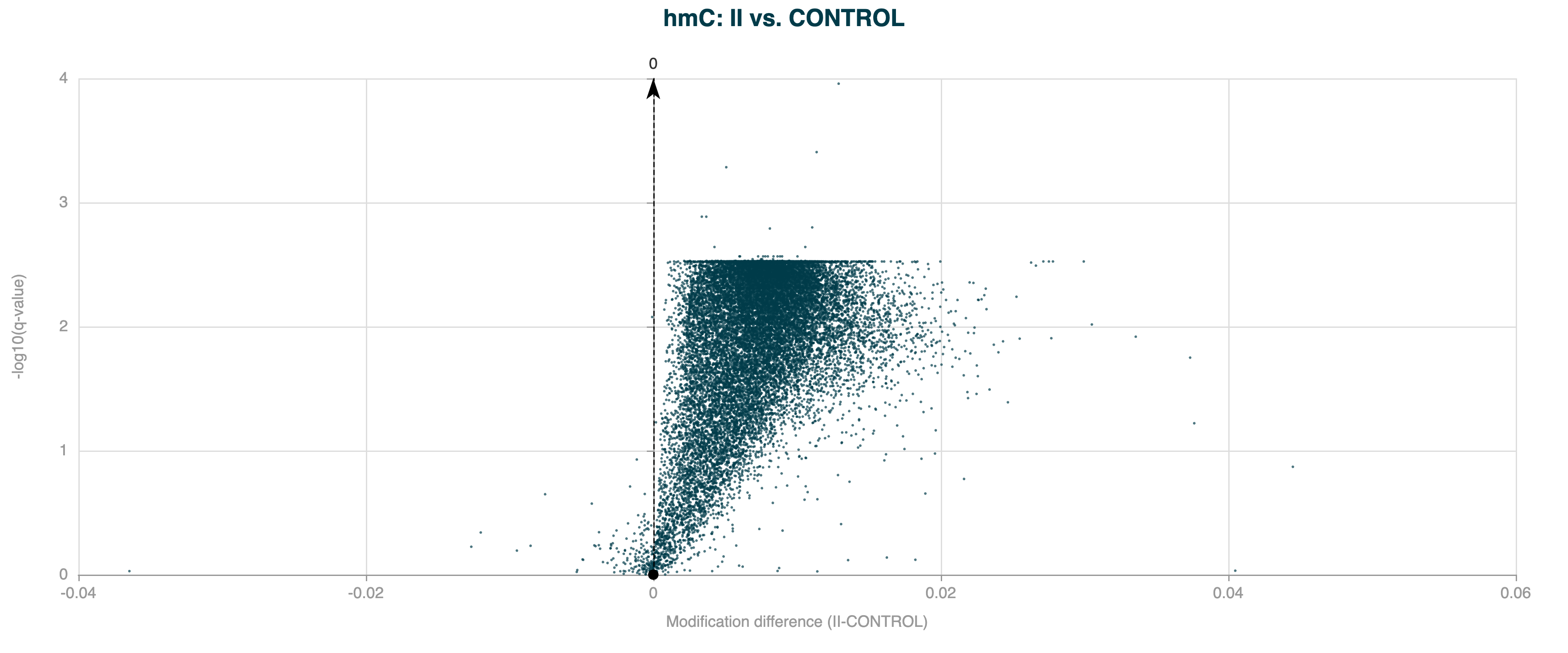
Figure 4 Volcano plot for 5hmC DMRs on gene bodies between Healthy Control and Stage II CRC cfDNA, showing many DMRs with similar q-values (plateau) after Benjamini-Hochberg correction.#
Here’s a breakdown of how the BH correction works and why multiple DMRs can have the same q-value:
Suppose we have m tests with raw p-values p1, p2, ... , pm:
First step of the BH is to sort them in ascending order, so that
p_sorted_1<p_sorted_2<...<p_sorted_m.Each sorted p-value gets a rank
i. The smallest p-value has rank1, the highest has rankm.The raw q-values are computed as
q_i = (p_sorted_i * m / rank_i).Crucially, the BH procedure forces that q-values are always increased with rank: to do that, replace each q-value with the minimum of itself and all q-values ranked below it.
Here’s how that might look in the data:
Rank |
Test |
p-value |
Raw q-value |
Adjusted q-value |
|---|---|---|---|---|
1 |
A |
0.0015 |
0.00750 |
0.00700 |
2 |
D |
0.0028 |
0.00700 |
0.00700 |
3 |
E |
0.0060 |
0.01000 |
0.01000 |
4 |
C |
0.0250 |
0.03125 |
0.03125 |
5 |
B |
0.0350 |
0.03500 |
0.03500 |
Notice how A has the lowest p-value, but the second lowest raw q-value? Therefore, the final step insures that the adjusted q-value of A is not higher than that of D by setting it to D.
This is not a bug, nor it is a problem - it’s what the BH correction does, which remains one of the most commonly used and most robust corrections available. This effect most likely arises when there are a lot of very small p-values. Therefore, it’s not surprising to observe this effect for gene-wide (i.e. very large region) p-values. Overdispersion correction can help mitigate this effect, but may not remove it completely. See Overdispersion correction for more details.
Overdispersion correction#
When calling DMRs, the modality dmr call tool can apply an overdispersion correction to the statistical model.
When calling DMRs, the modality dmr call tool uses a logistic regression model to
compare methylation levels between groups, assuming that methylation counts follow a binomial distribution
(i.e., the variance is determined by the mean). The aim is to test the null hypothesis i.e., that there is no difference in
5mc, 5hmC, or modC methylation between the groups.
The comparison results in a p-value for any difference. A p-value tells you the likelihood that the difference being observed is due to random noise. The smaller the p-value, the more likely the difference is a genuine effect, and the null hypothesis can be rejected.
p-values hold values between 0 and 1 and when plotted, you would expect to see a flat distribution across most values but with a higher value close to 0. The peak close to 0 should represent a slight increase in frequency above the rest of the p-value distribution.
In real-world methylation data, the observed variance is often greater than expected under a simple binomial model due to biological and technical variability. This is called overdispersion, and can lead to increased levels of false positives, seen as a much higher peak close to 0 than expected. In the example below, DMR calling has been applied to human gene bodies, which are prone to overdispersion due to the large region size.
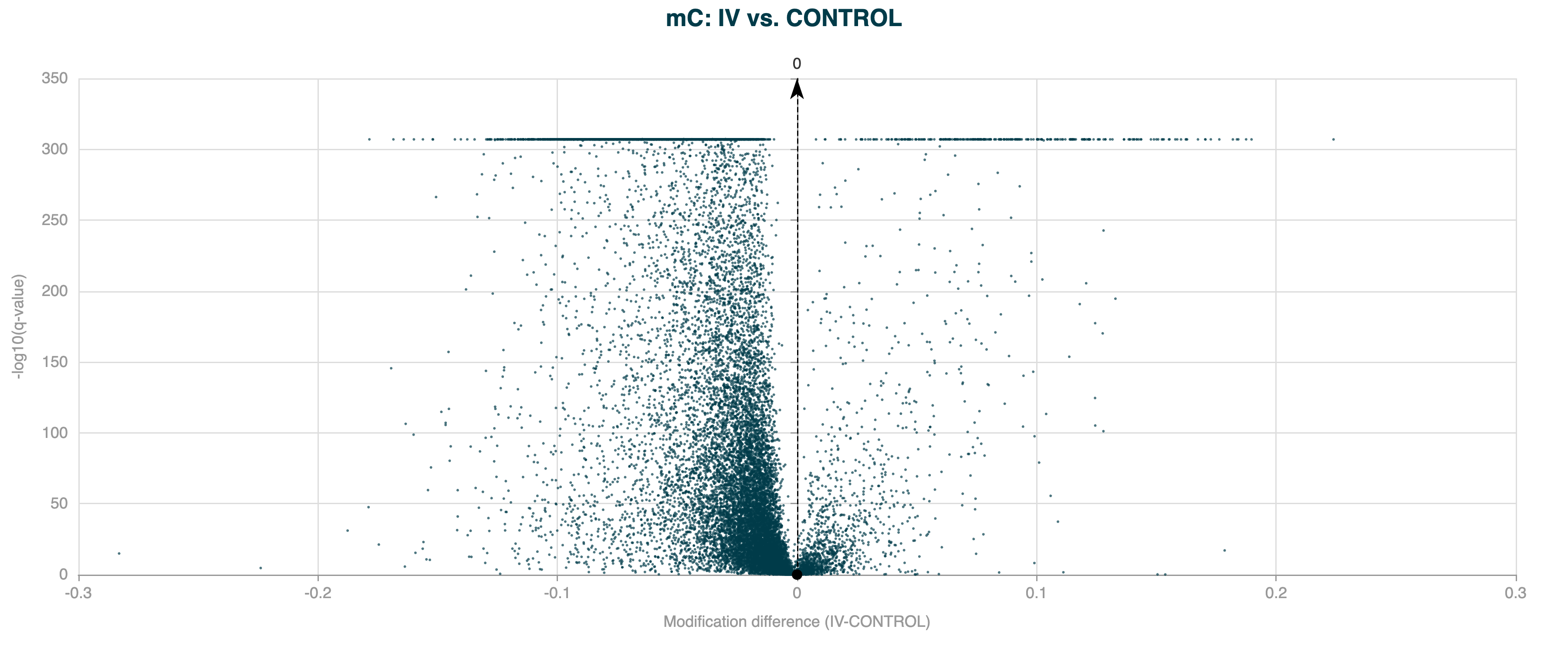
Figure 5 Volcano plot for 5hmC DMR results for human gene bodies, without overdispersion correction applied.#
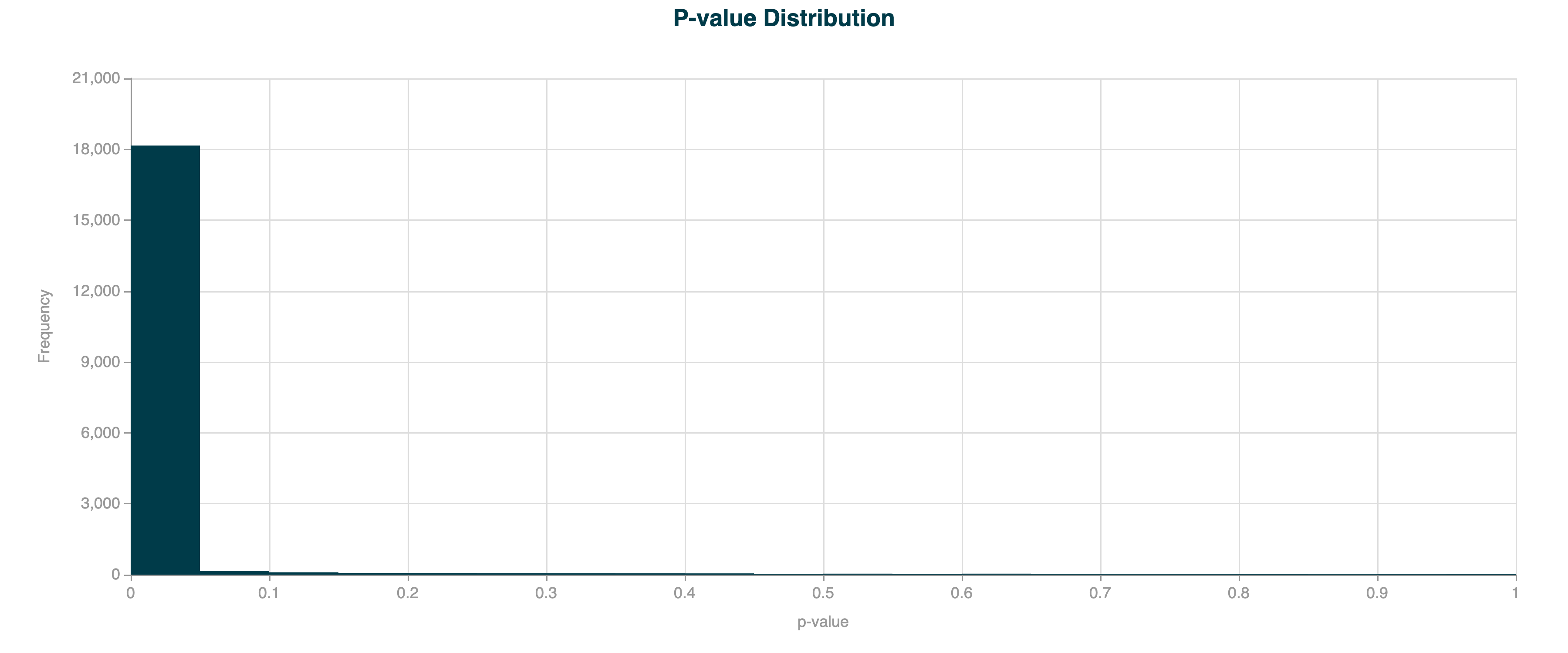
Figure 6 p-value distribution for 5hmC DMR results for human gene bodies, without overdispersion correction applied.#
When --overdispersion true is used, the model estimates an additional dispersion parameter (φ) for each region,
following the approach of McCullagh and Nelder (1989, Generalised Linear Models, Chapter 4.5).
This parameter adjusts the standard errors of the regression coefficients.
The Wald test statistic and p-values are then computed using these adjusted standard errors, providing more accurate significance estimates.
In the example below, the DMR calling has been applied to the same human gene bodies, but with overdispersion correction applied.
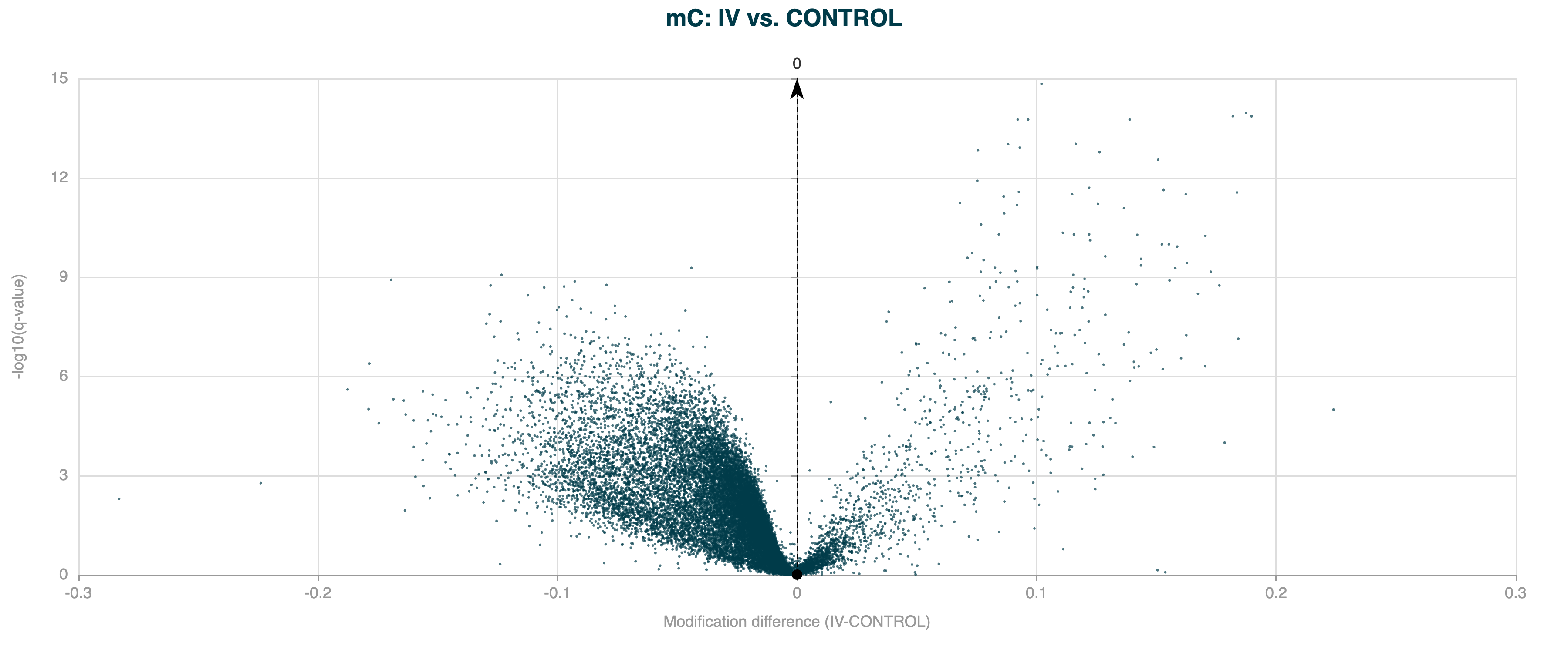
Figure 7 Volcano plot for 5hmC DMR results for human gene bodies, with overdispersion correction applied.#
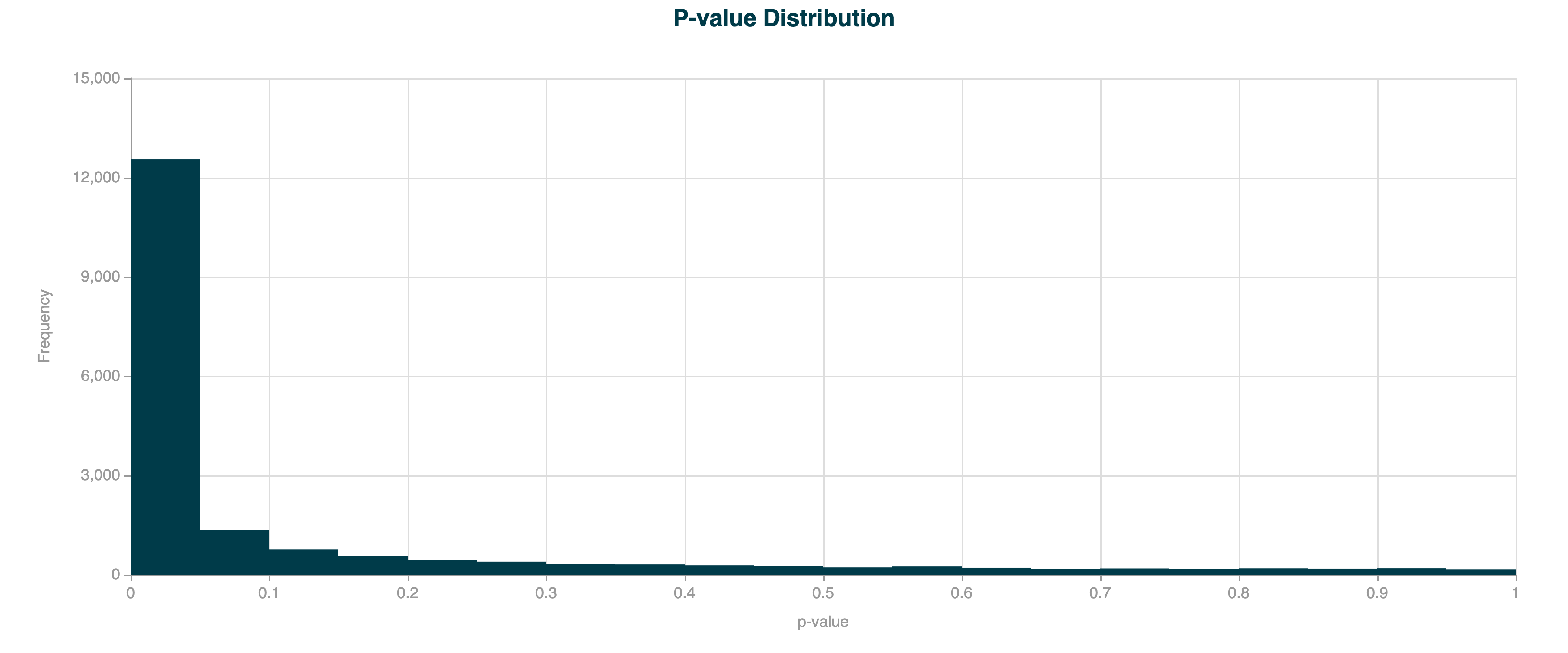
Figure 8 p-value distribution for 5hmC DMR results for human gene bodies, with overdispersion correction applied.#
Without overdispersion correction, the model can underestimate the true variance, leading to inflated test statistics and an excess of false positives (i.e., regions incorrectly identified as significant). Overdispersion correction ensures that statistical inference is robust, especially when analysing large regions, heterogeneous samples, or data with technical noise.
You can diagnose the need for this option by inspecting the p-value histogram from your DMR results: a very sharp peak near zero suggests overdispersion is present and correction may be needed.
Running both modes simultaneously#
If you are unsure whether overdispersion correction is appropriate for your data, you can use --overdispersion both.
This runs the DMR model twice — once with and once without overdispersion correction — in a single invocation.
Two BED result files are produced, distinguished by their suffixes:
*_without_od.bed— standard model (no correction)*_with_od.bed— overdispersion-corrected model
A single provenance JSON sidecar is written, and separate .ini track configuration files are generated for each result.
This allows you to compare the two sets of results side by side and decide which is more appropriate for your dataset.
Example:
modality dmr call \
--sample-sheet metadata.csv \
--zarr-path data.zarrz \
--condition-array-name label \
--overdispersion both
Sample size requirements for DMR analysis#
We have implemented automatic sample size validation to prevent unreliable statistical inference in DMR calling. Logistic regression requires sufficient samples per group to estimate within-group variability - with too few samples (such as one sample per group), the model perfectly fits the limited data points but produces unreliable coefficients and artificially small standard errors, leading to meaningless p-values and increased false positive rates. Each group must have more samples than the number of model parameters (1 + number of optional covariates like age or batch) to ensure valid statistical inference - for example, with no additional covariates, each group needs at least 2 samples, increasing to 3 samples per group when adding one covariate. When insufficient samples are detected, the algorithm automatically skips the statistical test and returns conservative p-values of 1 and test statistics of 0 for all regions, effectively preventing false positives, and logs a warning message to inform users of the sample size limitation.
Users with insufficient samples can still examine the biological effect sizes by looking at the methylation differences between groups (mod_difference column) and fold changes (mod_fold_change column) in the results, which remain meaningful descriptive statistics even when statistical significance cannot be reliably assessed.
Calling DMRs#
modality dmr call identifies regions where differentiated methylation occurs between different experimental groups.
Examples of these groups are disease and normal, treated or un-treated or time-course experiments.
A DMR is a defined genomic region where summed CpG sites show statistically significant differences in methylation between groups.
Analyses may be scanning the whole genome, divided into fixed windows, or focussed on specific annotated regions provided in BED format.
Inputs#
modality dmr call requires the following inputs:
Zarr store(s): Paths to each Zarr store to include in the analysis.
Sample sheet: Path to the sample sheet file containing metadata about the sample groups, or covariates. Covariates can be continuous (integers or floats) or categorical (strings).
Output directory (optional): Path to the output directory where the result files will be saved.
Command#
DMR calling is invoked by typing modality dmr call into the CLI terminal. To display a list of permitted arguments in the terminal, enter:
modality dmr call --help
Typical usage example:
modality dmr call \
--sample-sheet <path/to/metadata.csv> \
--zarr-path <path/to/your/data.zarrz> \
--condition-array-name <disease_stage> \
--methylation-contexts mc hmc \
--bedfile <path/to/regions.bed> \
--output-dir <path/to/output/directory>
The following table describes the tool options:
OPTION |
Required |
Description |
|---|---|---|
|
Yes |
Path to a sample sheet file
containing a column with the header
e.g., |
|
Yes |
One or more paths to Zarr files separated by spaces. e.g., |
|
Yes |
String for grouping samples by a
variable in the sample sheet, for
DMR calling. The
e.g., |
|
No |
String to specify the methylation contexts to be analysed. Multiple contexts can be specified by separating them with spaces. The available contexts are: If not specified, defaults are
auto-detected: e.g., |
|
No |
Integer to specify the window size
for DMR analysis. This option is
mutually exclusive with the
e.g., |
|
No |
String to specify the covariates to
be included in the analysis.
Multiple covariates can be specified,
which must match the column headers
in the e.g., |
|
No |
Path to a BED file containing regions
of interest for DMR analysis. Any
columns included past e.g., |
|
No |
Integer to specify the minimum
context sequencing depth for
filtering. Filters the positions if
the mean coverage across all samples
in each position is lower than
the desired filter value. Applied
before grouping by e.g., |
|
No |
Integer to filter out regions with fewer than the specified number of contexts. Only regions with at least this many contexts will be included in the results. Default is 0 (no filtering). e.g., |
|
No |
String to restrict analysis to a
specific genome region. Use the
format e.g., |
|
No |
Flag to disable strand merging to retain separate CpG sites on opposite strands (default: False, strands are merged). e.g., |
|
No |
Control the overdispersion correction
mode. Accepted values:
e.g., |
|
No |
This feature is optional but
recommended. If provided, the first
condition in the list will be used as
the reference (control) condition for
DMR analysis. All other conditions
will be compared to this condition,
and then compared to each other.
If not provided, sample order will
match the e.g., |
|
No |
Option to specify a path to an output directory. If not specified, the default output path is to the current working directory. e.g., |
|
No |
Option to specify whether to compress the DMR output file using gzip compression. Include this flag to turn ON compression. |
For more information on strand merging, see the Merging CpG strands section.
Important
The --window-size and --bedfile options are mutually exclusive.
You must specify either a window size for genome-wide tiling or provide a BED file with regions of interest for DMR analysis.
If neither option is provided, the tool will default to a window size of 1000 base pairs.
Outputs#
The modality dmr call tool generates the following outputs in the
specified output directory:
DMR result files: BED files containing the identified DMRs for each group pair and methylation context specified. The DMRs are annotated with the following columns:
Column |
Description |
|---|---|
|
Contig name. |
|
Start position. |
|
End position. |
|
Number of contexts in the window or region. |
|
Mean coverage of the contexts in the window or region. |
|
Mean methylation level for group 1 (e.g., control). |
|
Mean methylation level for group 2 (e.g., condition). |
|
Difference in methylation level between groups 1 and 2, where mean(group 2) / mean(group 1). Note: Specifying the |
|
Difference in methylation level between groups 1 and 2, where mean(group 2) - mean(group 1). Note: Specifying the |
|
The Wald test statistic associated with the p-value. |
|
The p-value of significance of the difference in methylation levels between the two groups. |
|
The FDR-corrected p-value. |
|
This is an optional column that would be
carried over from your BED file if provided
see |
|
This is an optional column that would be
carried over from your BED file if provided
see |
The DMR result files are named according to the following convention:
dmr_<group1>_<group2>_<context>_window_<window_size>bp.bed, or;dmr_<group1>_<group2>_<context>_bedfile.bedif a BED file is provided.
A DMR result file in BED3+ format will be generated per pairwise condition, per modification. For example, for a 6-base DMR analysis that includes both methylation contexts (mc and hmc), with a DMR sample sheet that contains three conditions (e.g. healthy, cancer_stage1, and cancer_stage4), six result BED files will be generated in the output directory.
Tip
Consider the use of --output-dir for efficient management of the output files.
.inifiles:
.ini file(s) contain the parameters used for the analysis, including the paths to input files.
The matched .ini files use the same naming convention as the DMR result files. There will be one .ini file per DMR result file.
The content of the .ini file can be changed, to control which plots are included when using modality tracks. This allows editing of figure titles, axis titles,
and optimising the order of the plots for clear communication of your findings. Navigate to the 1. Auto-generated .ini file section for more details.
Provenance metadata:
JSON sidecar files containing metadata about the analysis, including the command used, timestamp, version, and system information.
The provenance metadata files are named according to the following convention:
modality_metadata_<YYYYMMDD>_<HHMMSS>.json.
Analysis examples#
Example 1: DMR analysis on a single Zarr dataset for specific regions of interest#
modality dmr call \
--sample-sheet <path/to/metadata.csv> \
--zarr-path <path/to/your/data.zarrz> \
--condition-array-name <disease_stage> \
--methylation-contexts mc hmc \
--bedfile <path/to/regions.bed> \
--output-dir <path/to/output/directory>
Example 2: DMR analysis on a single Zarr dataset for genome-wide screening by window size#
modality dmr call \
--sample-sheet <path/to/metadata.csv> \
--zarr-path <path/to/your/data.zarrz> \
--condition-array-name <disease_stage> \
--methylation-contexts mc hmc \
--window-size 1000 \
--output-dir <path/to/output/directory>
Plotting differentially methylated regions (DMRs)#
The modality dmr plot tool generates visualisations to aid the interpretation of DMR analysis.
The visualisations are volcano plots and histograms of p-values for each DMR result file, providing insights into the significance and magnitude of methylation differences between groups. These visualisations are helpful for result filtering, identifying significant DMRs and assessing the overall quality of the analysis.
The outputs of dmr plot provide insights into:
The relationship between methylation differences and statistical significance: Volcano Plots
The distribution of p-values across all tested regions, as a QC for DMR calling: p-value Histograms
Note
For different experimental reasons, there can be a very high frequency of p-values close to ‘0’,
if this is the case, refer to Overdispersion correction for additional details regarding the --overdispersion option available in modality dmr call.
Inputs#
modality dmr plot requires the following inputs:
DMR result file(s): Paths to the output(s) of the
modality dmr calltool, in.bedformat. These files contain the DMRs identified in the analysis.Output directory (recommended): Path to the output directory where the result files will be saved.
Command#
The DMR visualisation tool is invoked by typing modality dmr plot into the CLI terminal. To display a list of permitted arguments in the terminal, enter:
modality dmr plot --help
Typical usage example:
modality dmr plot \
--dmr-results <path/to/dmr_results.bed> \
--filter-qvalue 0.05 \
--filter-mod-difference 0.1 \
--output-dir <path/to/output/directory>
The following table describes the tool options:
OPTION |
Required |
Description |
|---|---|---|
|
Yes |
One or more paths to DMR result files.
These files are generated
by the e.g., |
|
No |
Float to filter DMRs by q-value. Only
DMRs with q-values e.g., |
|
No |
Float to filter DMRs by modification
difference. Only DMRs with
e.g., |
|
No |
Directory to save the generated plots. If not specified, the default output path is the current working directory. e.g., |
Outputs#
The modality dmr plot tool generates an HTML report per DMR results file input, in the specified output directory. Each HTML report contains the following sections:
DMR summary table
Confirms the two Group names included in the DMR analysis, and which is the designated Control group and Test group, for calculating the modification difference, and directionality of hypo- and hyper-methylation.
Shows the total number of regions (or windows) tested (after depth filtering).
Shows the mean context depth, of all regions (or windows) passing q-value and/or mod-difference filter, if applied.
Shows the mean number of contexts in all regions (or windows) passing q-value and/or mod-difference filter, if applied.
If filtering was applied, the table will also show the number (and %) of regions (or windows) passing q-value and/or mod-difference filter.
Volcano plot
Visualise the relationship between methylation differences (x-axis) and log transformed statistical significance \(-\log_{10}(\text{q-value})\) (y-axis).
DMRs are included based on the specified q-value and modification difference thresholds.
If provided with a
--bedfilewith multiple values in theAnnotationcolumn, the volcano plot data points will be colour-coded and an Annotation legend will be shown on the plot. Note that input bedfile annotations are carried over to the DMR results file used for plotting.The volcano plot is interactive, allowing you to hover over points to see details about individual DMRs.
The plot can be manipulated with embedded tools, such as zooming and panning, to focus on specific regions of interest.
Use the chart tools to save the image as
.png.
p-value histogram
Shows the distribution of p-values across all tested regions.
Provides an overview of the statistical significance of the results.
Helps to assess the overall quality of the DMR calling process, namely the proportion of significant DMRs.
If provided with a
--bedfilewith multiple values in theAnnotationcolumn, a stacked histogram will be shown together with a legend on the plot. Note that input bedfile annotations are carried over to the DMR results file used for plotting.
The output files are named according to the following convention:
HTML report:
<dmr_result_file>_volcano.html
Analysis examples#
Example 1: Plotting DMR results with filters#
modality dmr plot \
--dmr-results <path/to/dmr_results.bed> \
--filter-qvalue 0.05 \
--filter-mod-difference 0.1 \
--output-dir <path/to/output>
Example 2: Plotting multiple DMR result files#
modality dmr plot \
--dmr-results <path/to/dmr1.bed> <path/to/dmr2.bed> \
--output-dir <path/to/output>
Generate genomic track plots#
The modality tracks tool generates genomic track plots for visualising methylation data, differentially methylated regions (DMRs), and gene annotations.
This tool uses an .ini configuration file to define the tracks to be plotted, allowing you to customise the visualisation according to their needs. The .ini file is viewed, modified or created in a standard text editor.
The outputs of modality tracks provide insights into:
Methylation traces: Visualising methylation levels across genomic regions for individual samples or groups.
Methylation differences: Highlighting differences in methylation levels between groups, specifically hyper- and hypo-methylation.
DMR tracks: Displaying the location and significance of DMRs.
Annotations: Adding biological context to the visualisation using GTF/GFF files.
These visualisations are essential for interpreting methylation data and understanding its biological relevance.
The modality tracks command will generate a single track plot (.png or .pdf) containing the individual tracks that are specified in
the .ini file.
The tracks in the track plot will appear in the order they are listed in the .ini file.
Inputs#
modality tracks requires the following inputs:
ini file: Path to the
.inifile to be included in the analysis.Output directory (optional): Path to the output directory where the result files will be saved.
This section details the data held in the .ini file, how it is structured, and which fields are editable.
Instructions for creating a specific new .ini file are provided and these can be saved for future use.
1. Auto-generated .ini file#
An .ini file is automatically generated by the modality dmr call tool when DMRs are called
and saved in the output directory --output-dir specified in the command.
The .ini file is divided into sections. A section is indicated by square brackets [ ], and is descriptive if the track type.
The use of mc/hmc/modC is dependent on the type of DMR analysis that was run.
The main sections are:
Section |
The section purpose |
|---|---|
|
Settings for each of the tracks e.g. size,title |
|
Methylation trace track for 5mC, 5hmC, or modC |
|
Methylation difference track for 5mC, 5hmC, or modC |
|
DMR bar track |
|
Adds vertical space between tracks |
|
Annotation track |
|
x-axis configuration |
Each of these sections has a set of components which are populated in the auto-generated .ini file.
An example .ini file, after running modality dmr call for hmc DMRs between two conditions, CONTROL and I
(specified by the column header, label, in the sample sheet metadata file:)
[DEFAULT]
width = 20
figure_title = CONTROL vs I - hmc DMRs
disable_strand_merging = False
min_coverage = 0
[mc methylation trace]
type = zarr_methylation_trace
zarr = path/to/data.zarrz
metadata = path/to/metadata.csv
ymin = 0
ymax = 1
height = 3
title = hmc fraction
modification_type = hmc
group = label
group_trace = True
group_points = True
group_names = CONTROL; I
[spacer]
[mc methylation diff]
type = methylation_diff
zarr = path/to/data.zarrz
metadata = path/to/metadata.csv
height = 3
title = hmc difference
modification_type = hmc
ymin = -0.6
ymax = 0.6
group = label
group_names = CONTROL; I
[spacer]
[dmr]
type = dmr_bar
title = DMR magnitude
file = path/to/DMR_hmc_CONTROL__I.bed
dmr_header_present = True
y_min = -0.5
y_max = 0.5
height = 2
[spacer]
;[gene]
;type = gtf_gff
;file = path/to/gencode.gff3.gz
;file_type = gff3
;title = gencode
;height = 2
[x-axis]
Note
The [gene] section is commented out with a semicolon at the start of the line, meaning that the gene annotation track will not be included in the plot. To include the gene annotation track, remove the semicolon at the start of the line and ensure that the file path and type are correct. You will need to ensure that the GTF/GFF file is available and correctly formatted for the track to be built successfully.
Note
Users can choose whether they want to use a GFF3 or GTF file for annotation. The default expects a GFF3 but to pass a GTF file instead, change from ;file_type = gff3 to ;file_type = gtf and exclude the semicolon “;”.
Each section has its own components, the next table explains the purpose of the component, which are required, default settings and where input is needed.
Details of [DEFAULT]
Component |
Setting |
Purpose |
|---|---|---|
|
default: 20 |
Width of the figure in inches |
|
default: None |
Title for the entire figure |
|
default: matches DMR call setting |
Boolean flag to disable CpG strand merging across all methylation tracks |
|
default: 0 |
Minimum coverage filter: removes positions with coverage < min_coverage from tracks. Note: fractions are calculated with built-in min_coverage=1, so 0-coverage positions are not plotted regardless of this setting. |
Details of [mc/hmc/modc methylation trace]
Component |
Setting |
Purpose |
|---|---|---|
|
required |
Must be |
|
required |
Path to the Zarr store |
|
required |
Path to the sample metadata CSV or TSV |
|
default: 0 |
Minimum value for y-axis |
|
default: 1 |
Maximum value for y-axis |
|
default: 3 |
Height of the track in inches |
|
default: None |
Title for the track |
|
required |
|
|
default: None |
Metadata column to group samples |
|
default: True |
Plot group traces (True/False) |
|
default: True |
Plot individual sample points (True/False) |
|
default: None |
Semicolon-separated group names, e.g., |
Details of [mc/hmc/modc methylation diff]
Component |
Setting |
Purpose |
|---|---|---|
|
required |
Must be |
|
required |
Path to the Zarr store |
|
required |
Path to the sample metadata CSV or TSV |
|
default: -0.6 |
Minimum value for y-axis |
|
default: 0.6 |
Maximum value for y-axis |
|
default: 3 |
Height of the track in inches |
|
default: None |
Title for the track |
|
required |
|
|
default: None |
Metadata column to group samples |
|
default: None |
Semicolon-separated group names, e.g., |
Details of [dmr]
Component |
Setting |
Purpose |
|---|---|---|
|
required |
Must be |
|
default: None |
Title for the DMR bar track |
|
required |
Path to the DMR BED file |
|
default: TRUE |
Whether the BED file has a header |
|
default: -0.5 |
Minimum value for y-axis |
|
default: 0.5 |
Maximum value for y-axis |
|
default: 2 |
Height of the track in inches |
Details of [gene]
Important
To enable the gene annotation track, remove the semicolon at the start of only the following lines in the .ini file, including in front of the [gene] section header. Ensure that the GTF/GFF file is available and correctly formatted for the track to be built successfully.
Component |
Setting |
Purpose |
|---|---|---|
|
required |
Must be |
|
default: None |
Title for the annotation track |
|
required |
Path to the GTF/GFF annotation file |
|
required |
|
|
default: 2 |
Height of the track in inches |
Details of [spacer]
There are no parameters for this section, when between sections it adds a vertical space between the tracks.
Details of [x-axis]
This is used to configure the x-axis and typically remains empty to deliver the default settings.
2. Customising .ini files#
The default modality tracks visual is auto-generated but it can be customised. Table A shows default settings and ranges for components that can be used and Table B focusses on modifications of the .ini file.
Changes are made by opening the .ini file in a text editor, updating with the changes and saving the file.
Important
Remember to save the updated .ini file and its path.
Table A: Default ranges
Component |
Default |
Range/Notes |
|---|---|---|
width |
20 |
>0 |
figure_title |
None |
Any string |
disable_strand_merging |
False |
True/False |
min_coverage |
0 |
>=0, track filtering (see Note) |
ymin, y_min |
0/-0.5/-0.6 |
Any float, typically -1 to 1 |
ymax, y_max |
1/0.5/0.6 |
Any float, typically -1 to 1 |
height |
2 or 3 |
>0 |
group_trace |
True |
True/False |
group_points |
True |
True/False |
group_names |
None |
Semicolon-separated, e.g., |
dmr_header_present |
True |
True/False |
Note
Understanding min_coverage in tracks:
There are two distinct coverage thresholds in the methylation plotting pipeline:
Fraction calculation threshold (always
min_coverage=1): When methylation fractions are computed (e.g., mc / total_c), positions with coverage < 1 are automatically set to NaN to avoid division by zero. This happens internally and cannot be changed.Track filtering threshold (default
min_coverage=0): This is the parameter you set in the.inifile. It filters positions AFTER fractions are calculated:min_coverage=0(default): No filtering - all positions are kept, including those with NaN fractions from 0 coverage (these appear as gaps in plots)min_coverage=N: Removes positions where coverage < N before plotting
Example: If you set min_coverage=10, positions with coverage 0-9 will be removed
from the plot. Positions with coverage ≥10 will be displayed with their methylation
fraction values.
Table B: modifying the ``.ini`` file
Modification |
How to modify |
|---|---|
Add tracks (plots) |
Copy and paste the section to duplicate the type of plot. |
Remove tracks (plots) |
Delete the section from the |
Change axis limits |
Adjust |
Change grouping |
Set |
Add gene annotation |
Remove |
Customise titles |
Edit |
Adjust panel order |
Rearrange the complete sections in the |
Use p-value |
Set |
3. Creating an .ini file from scratch#
When neither the default nor modification options for .ini file deliver the format of results required, a specific .ini file can be built. Using the details above as guidance for required components, here are the steps to building an .ini file:
Open a plain text editor such as TextEdit or a code editor such as Visual Studio Code.
Define the sections remembering they need to be enclosed in square brackets [Section name].
Add the key-value pairs e.g. title=hmc difference. The key is the ‘title’ and the value is ‘hmc difference’. The key-value pairs are therefore the components and values required to create the plot required.
Save the file with an
.iniextension (e.g., config.ini) and note the path for use withmodality tracks.
Important
There are best practices when working with .ini files:
Always check that the file paths are correct and accessible.
Use consistent group names with the sample sheet.
Consider the y-axis scale when comparing mC and hmC DMRs.
Save custom
.inifiles to be able to re-use the format.
Command#
The tracks visualisation tool is invoked by typing modality tracks into the CLI terminal. To display a list of permitted arguments in the terminal, enter:
modality tracks --help
Typical usage example:
modality tracks \
--tracks-file <path/to/config.ini> \
--region chr1:100000-200000 \
--output-dir <path/to/output>
The following table describes the tool options:
OPTION |
Required |
Description |
|---|---|---|
|
Yes |
Path to the track configuration file
(.ini) that defines the tracks to be
plotted. The e.g., |
|
Yes |
Genomic region to plot. Use the
format e.g., |
|
No |
Path to save the generated plot. The output file format is determined by the file extension (e.g., .png, .pdf). e.g.,
|
|
No |
Format for the output image file. Options: ‘png’, ‘jpg’, ‘pdf’, ‘svg’. Default: ‘png’. e.g., |
|
No |
Integer to specify the number of base pairs to extend the region on either side. Effectively, to zoom out. e.g., |
|
No |
Flag to disable strand merging to retain separate CpG sites on opposite strands (default: False, strands are merged). e.g., |
For more information on strand merging, see the Merging CpG strands section.
Outputs#
The modality tracks tool generates a genomic track plot in the specified output file format (e.g., .png or .pdf). The plot includes the following tracks, as defined in the configuration file:
Methylation trace:
Visualise methylation levels across the specified genomic region.
Grouped by sample condition, as defined in the metadata file.
Modification difference:
Highlight differences in methylation levels between groups.
Smoothed and raw differences are displayed.
The solid line in the methylation trace and methylation difference tracks provides a smoothed representation of the data for improved readability. Smoothing is performed using a coverage-weighted Gaussian kernel-based approach. The smoothing scale is adaptive based on the length and density of the region that is being plotted.
DMR track:
Display the location and significance of DMRs.
Positive and negative DMRs are colour-coded.
Uses q-values by default.
Annotation:
Add biological context using GTF/GFF files.
Display gene names, exons, and other features.
The output file is named according to the specified path and file extension, e.g., track_plot.png.
Analysis examples#
Example 1: Generating a track plot for a specific region#
modality tracks \
--tracks-file <path/to/config.ini> \
--region chr1:100000-200000 \
--output-dir <path/to/output>
Example 2: Adding padding to the genomic region#
modality tracks \
--tracks-file <path/to/config.ini> \
--region chr1:100000-200000 \
--padding 1000 \
--output-dir <path/to/output>
Extracting methylation statistics#
The modality get command is designed to extract methylation statistic summaries for genomic regions or windows, from methylation data stored in Zarr format, from the duet software.
This analysis can be used for creating datasets for classification algorithms, modelling gene expression, and examining potential coverage or methylation biases.
This tool supports four key statistical summaries under the modality get command:
Subcommand |
Description |
Example Use Case |
|---|---|---|
|
Counts the number of methylation contexts in each region. |
Identify the number of methylation context in each region or window, to allow filtering or preparation of lists ahead of running other analyses. |
|
Computes the sum
of values for
selected
|
Summarises total modifications across a region. |
|
Computes the
mean value for
selected
|
Assess mean coverage across a region. |
|
Computes the
fraction for
selected
|
Calculate the fraction of a field over
|
The four subcommands provide a specific type of statistical analysis, enabling you to explore methylation patterns across genomic regions of interest.
The modality get command serves two main purposes:
Extraction of methylation statistics into a
tsv.gzfile, enabling feature-set generation and downstream analysis.Visualise methylation statistics in HTML reports, to explore the distribution, correlation and hierarchical clustering of methylation across regions.
A typical Feature Extraction workflow requires:
Specified data input.
Selection of a statistical operation and methylation context(s) of interest.
Specified genomic region(s) of interest or fixed window size.
Which in turn provides:
Summarised methylation data as
tsv.gz.HTML reports with interactive plots that can be saved as
.png, formeanandregional-fracsubcommands.
The next three sections explain how to extract the DNA methylation statistics, create and interpret HTML visualisations for insight into patterns in DNA methylation.
Inputs#
The modality get command requires the following inputs:
Zarr path: Path to the Zarr file(s) containing methylation data.
Fields: Type(s) of cytosine to extract. Allowed
--fieldsvary by subcommand (sum,mean, andregional-frac).Region definition: Either a BED3+ file defining multiple genomic regions of interest, a single specified region, or a window size for genome-wide analysis.
Note
Field values: See Zarr store section for more information about available field values.
Optional input files#
Optional file inputs are determined by the analysis being carried out:
A text file in BED3+ format, listing the specific region(s) of interest.
A text file in CSV or TSV format, containing sample group or condition metadata.
Commands#
The modality get command is invoked by typing modality get into the CLI terminal.
The command format is modality get <subcommand> <OPTIONS>.
Subcommands#
To display a list of permitted arguments for a specific subcommand, type:
modality get <subcommand> --help
# e.g., for the `mean` subcommand
modality get mean --help
# e.g., for the `count` subcommand
modality get count --help
# e.g., for the `sum` subcommand
modality get sum --help
# e.g., for the `regional-frac` subcommand
modality get regional-frac --help
Example usage:#
This illustration shows the subcommand with instructions to:
Bring in specific Zarr store and .bed files
Visualise the results in violin, pearson correlation and regional heatmap plots
Save the output files (Data
.tsv.gzand Visualisations.html) to a specific output folder
modality get regional-frac \
--zarr-path <path/to/data.zarrz> \
--bedfile <path/to/regions.bed> \
--heatmap \
--output-dir <path/to/output>
Common options#
The following table describes the common options available. Note that not all options are available for all subcommands:
Option |
Required |
Description |
|---|---|---|
|
Yes |
Path to the Zarr file(s) to be analysed. e.g., |
|
Conditional |
Type(s) of cytosine to extract. Allowed fields vary by subcommand. Optional for Auto-detection ( Required ( e.g., |
|
No |
Integer to specify the minimum context sequencing depth for filtering. Filters the positions if the mean coverage across all samples in each position is lower than the desired filter value. Applied before aggregation with e.g., |
|
No |
Integer to filter out regions with fewer than the specified number of contexts. Only regions with at least this many contexts will be included in the results. Default is 0 (no filtering). e.g., |
|
No |
Path to a BED file defining genomic regions of interest. Mutually exclusive with e.g., |
|
No |
Integer to specify the window size for analysis. Mutually exclusive with e.g., |
|
No |
String to restrict analysis to a specific genome region. Use the format e.g., |
|
No |
Path to a CSV file containing sample metadata. e.g., |
|
No |
Optional group name for aggregation. Reduces the dataset by summing count values for samples within each group. This affects all downstream analysis (correlation, PCA, etc.). Use this for merging technical replicates or combining samples by condition. Requires |
|
No |
Column name in the sample sheet to use for ordering samples in plots. Samples will be sorted by this group for visual organisation only. Does not perform aggregation - all individual samples remain in the analysis. Requires |
|
No |
Flag to include a regional heatmap in the HTML report. Only available for |
|
No |
Directory to save the output files. Default: current working directory. e.g., |
|
No |
Flag to turn ON case-sensitivity for e.g., |
|
No |
Compress output files using bgzip and create Tabix index. Requires installation of non-python dependencies (see Non-python dependencies). e.g., |
|
No |
Flag to disable strand merging to retain separate CpG sites on opposite strands (default: False, strands are merged). e.g., |
For more information on strand merging, see the Merging CpG strands section.
HTML report visualisations#
Important
The HTML report is generated with get mean and get regional-frac commands, containing violin and Pearson correlation plots for each modification type 5mC, 5hmC, 5modC.
If specified, a regional heatmap is also included for regional-frac and mean.
Plotting is not supported for count and sum subcommands, as these operations do not produce meaningful visualisations.
HTML files include multiple plots, with tools for performing basic manipulations such as saving the image in PNG format. A description of supported plots is provided below:
Violin: A violin plot visually represents the density of data across a range of values. It will show DNA modification distribution across the DNA region. The violin also includes box and whisker plots.
Correlation: A Pearson correlation matrix plot which visualises the relationship between two or more variables. It does not show causality. The deeper the colour the higher the correlation.
Regional heatmap: A heatmap visualising the correlation and heirarchical clustering of methylation profiles across genomic regions and samples.
Optimising plots#
Plotting subsets of samples
When working with a large number of samples, it may be useful to restrict the plotting to specific samples or group the analysis output based on sample labels. This may be for speed or where some samples have a specific biological element that is of interest.
The following options apply to plotting functions:
--aggregate-by-group: Aggregates samples based on a common ID or group name column in the--sample-sheetwhere group labels are listed e.g. control or condition 1, treated or untreated, healthy or disease stage 1, etc. This action will reduce output results files and plots.--order-by-group: Orders samples based on a specific column in the--sample-sheetwhere group labels are listed. This action will not aggregate samples but will order them on the plot axes for easier visual interpretation. Result files are not reduced.--case-sensitive: This option allows for case-sensitive group labels, negating the default behaviour of converting group labels to uppercase in plots (see below).
Group labels
For plotting, the group label values are converted to uppercase by default. For example, Healthy and healthy would be assigned to the same group, HEALTHY.
To override this behaviour, use the --case-sensitive option to report Healthy and healthy as separate groups.
Outputs#
According to the modality get subcommand and analysis performed, the software shall return the following result files to the output directory.
If an --output-dir path is not provided, the result files shall be saved in the current working directory:
Compressed TSV result file(s):
Contains the extracted statistics for each genomic region.
Columns include genomic coordinates, extracted values, and additional metadata in BED format.
The file name is based on the subcommand used, e.g.,
<methylation-context>_<subcommand>.tsv.gz.
HTML report:
Visualise the extracted statistics.
Saved in the specified output directory as HTML files.
Provenance metadata:
JSON sidecar files containing metadata about the analysis, including the command used, timestamp, version, and system information.
The provenance metadata files are named according to the following convention:
modality_metadata_<YYYYMMDD>_<HHMMSS>.json.The metadata includes the command used, timestamp, version, and system information.
Analysis examples#
Example 1: Counting contexts in genomic regions
This operation counts the number of cytosine contexts in each region.
The --fields argument is optional (defaults to c), as the count is identical regardless of which field is specified.
It can also be used in conjunction with a minimum coverage setting.
modality get count \
--zarr-path <path/to/data.zarrz> \
--bedfile <path/to/regions.bed> \
--output-dir <path/to/output>
Example 2: Summing methylation counts across regions
This operation sums the total number of modifications across each region.
For 6-base datasets, mc and hmc fields are auto-detected; for 5-base datasets, modc is auto-detected.
You can override by explicitly specifying --fields.
modality get sum \
--zarr-path <path/to/data.zarrz> \
--bedfile <path/to/regions.bed> \
--output-dir <path/to/output>
Example 3: Calculating mean context value in each region
This operation can be used to assess the average coverage per CpG.
For example, average number of CpGs covered at a specific depth in sequencing (total), or with a cytosine basecall (total_c).
modality get mean \
--zarr-path <path/to/data.zarrz> \
--bedfile <path/to/regions.bed> \
--fields total_c \
--output-dir <path/to/output>
Example 4: Computing regional methylation fraction over total_c
This operation is suitable for methylation fraction analysis and will calculate the proportion
of methylation out of the total number of unmodified and modified Cs (total_c), for each genomic region or window.
For 6-base datasets, mc and hmc fields are auto-detected; for 5-base datasets, modc is auto-detected.
You can override by explicitly specifying --fields.
modality get regional-frac \
--zarr-path <path/to/data.zarrz> \
--bedfile <path/to/regions.bed> \
--heatmap \
--output-dir <path/to/output>
Audit trail#
Terminal feedback#
modality XPLR provides immediate feedback in the terminal when commands are executed, showing:
The command being run with all specified parameters
Progress indicators for long-running operations
Confirmation of output file creation
Summary information upon successful completion
This feedback helps to confirm the command was interpreted correctly and analysis progression is being tracked.
Below is an example of the terminal output and provenance feedback generated during a typical modality biological-qc run:
2025-06-19 12:15:19 Provenance information:
2025-06-19 12:15:19 cmd='path/to/env/bin/modality biological-qc --zarr-path path/to/data.zarrz'
timestamp=2025-06-19T12:15:17.997570
version=1.0.0
user=user_name
2025-06-19 12:15:19 Running biological QC analysis:
2025-06-19 12:15:19 Zarr path(s): ['path/to/data.zarrz']
2025-06-19 12:15:19 Region: None
2025-06-19 12:15:19 Sample Sheet: None
2025-06-19 12:15:19 Group Name: None
2025-06-19 12:15:19 Output Dir: path/to/output
2025-06-19 12:16:27 | WARNING | path/to/env/lib/python3.x/site-packages/dask/core.py:127: RuntimeWarning: invalid value encountered in divide
return func(*(_execute_task(a, cache) for a in args))
2025-06-19 12:16:49 | WARNING | path/to/env/lib/python3.x/site-packages/dask/core.py:127: RuntimeWarning: invalid value encountered in divide
return func(*(_execute_task(a, cache) for a in args))
2025-06-19 12:16:55 Biological QC report generated: path/to/output/BioQC_YYYYMMDD_HHMMSS/biological_qc_report_<n>_samples_YYYYMMDD_HHMMSS.html
2025-06-19 12:16:55 Wrote provenance sidecar file to path/to/output/BioQC_YYYYMMDD_HHMMSS/modality_metadata_YYYYMMDD_HHMMSS.json
Logging and provenance tracking#
Logs for every CLI command are written to a file named modality_cli.log in the current working directory for further inspection.
Logging provides a record of events during the execution of modality XPLR commands. It helps to:
Debug issues by reviewing detailed logs.
Monitor the progress of long-running commands.
Maintain a history of executed commands for reproducibility.
Provenance tracking ensures that every analysis is traceable by recording:
The command executed.
The timestamp of execution.
The working directory and user.
The version of modality XPLR used.
System information (e.g., OS, Python version).
A list of output files generated.
This metadata is critical for reproducibility and for understanding the context of results.
Process#
Provenance metadata: Captured automatically for every command.
Output registration: All output files are registered and tracked.
Sidecar file: Provenance metadata is saved in a JSON sidecar file alongside outputs.
File comments: Metadata can be embedded as comments in output files (e.g. BED files).
For example, provenance metadata includes:
Command: modality dmr –sample-sheet metadata.tsv –zarr-path file.zarr
Timestamp: 2025-05-20T12:00:00
modality XPLR version: 1.0.0
User: first_name.last_name
System info: { “os”: “macOS”, “python_version”: “3.11” }
Outputs: [ “path/to/output/dmr_results.bed” ]
Provenance data is written to a JSON sidecar file in the output directory. A timestamp is included in the filename.
e.g. modality_metadata_<YYYYMMDD>_<HHMMSS>.json
For CLI commands that produce BED format result files, the provenance metadata can be embedded as comment header lines in the output file.
Setting verbosity levels#
How to set verbosity levels#
The verbosity level in modality commands controls the amount of information displayed in the terminal during execution.
This feature is useful for debugging, monitoring progress, or reducing unnecessary output.
modality XPLR supports three verbosity levels:
Default (no flag): Displays warnings and errors and command feedback only.
Information: Use
--verboseor-vto display informational messages, warnings, and errors.Debug: Use
-vvto display detailed debugging information, along with informational messages, warnings, and errors.
To set the verbosity level, use the --verbose, -v (Information) or -vv (Debug) flags when running a modality command.
The flag must be placed after the command and before any other options.
Each additional v increases the verbosity level. For example:
modality -v biological-qc \
--zarr-path <path/to/data.zarrz> \
--output-dir <path/to/output/directory>
modality -vv get regional-frac \
--zarr-path <path/to/data.zarrz> \
--fields mc hmc \
--bedfile <path/to/regions.bed> \
This tool is particularly useful for monitoring long-running commands such as modality dmr call. Setting the verbosity level to Information
will display progress bars, helping you to understand how far along the process is and if any issues arise. Below is an example
of the terminal output when running modality -v dmr call:
Figure 9 Terminal output showing progress bars for modality -v dmr call command.#
Managing configuration#
The modality config command group provides tools for viewing and updating modality XPLR settings.
Telemetry consent#
modality XPLR collects anonymous usage events to help improve the tool. On first run, you will be asked once whether you would like to share these events with biomodal. Your preference is saved to ~/.modality_xplr/config.yaml and you will not be asked again.
You can view or change your preference at any time using the modality config telemetry command.
View current setting#
Run without flags to display your current telemetry preference:
modality config telemetry
Example output when sharing is enabled:
Telemetry sharing is currently enabled.
Example output when sharing is disabled:
Telemetry sharing is currently disabled.
Enable sharing#
modality config telemetry --enable
Disable sharing#
modality config telemetry --disable
Note
Changes to your telemetry preference take effect on the next command invocation, at which point an audit record is sent to biomodal to confirm the update.